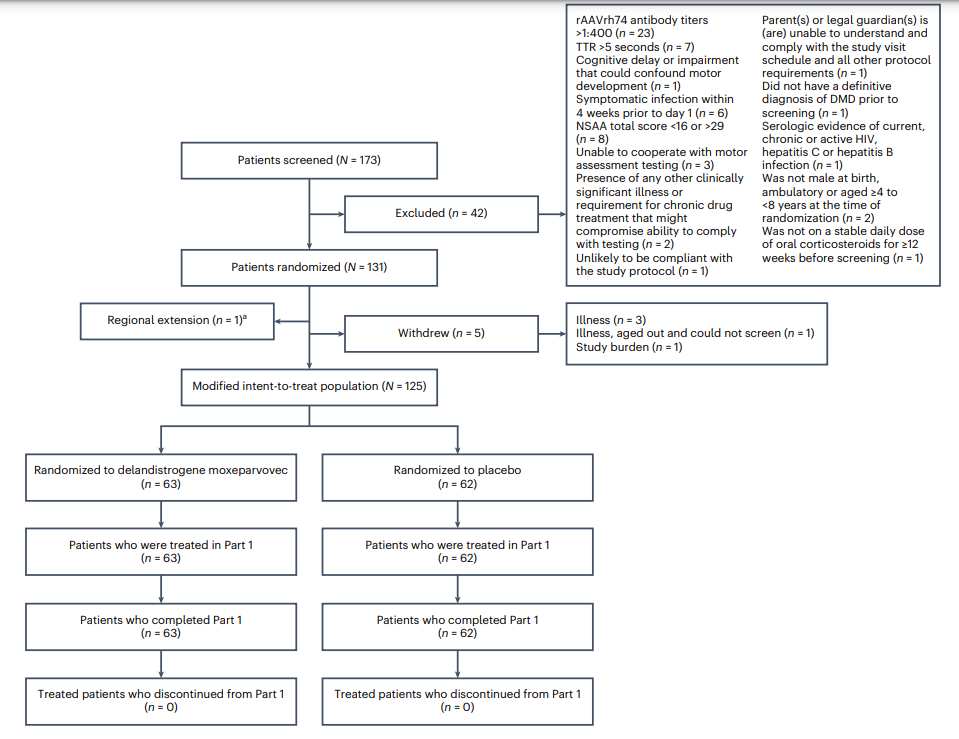
The paper presents findings from a phase 3 clinical trial (EMBARK) evaluating the efficacy and safety of a gene therapy, delandistrogene moxeparvovec, in patients with Duchenne muscular dystrophy (DMD). DMD is a severe, inherited condition that progressively weakens muscles due to the absence of dystrophin. The therapy uses an AAV vector to deliver a functional dystrophin gene into muscle cells. The trial involved boys aged 4 to 8, who were randomized to receive either the therapy or a placebo.
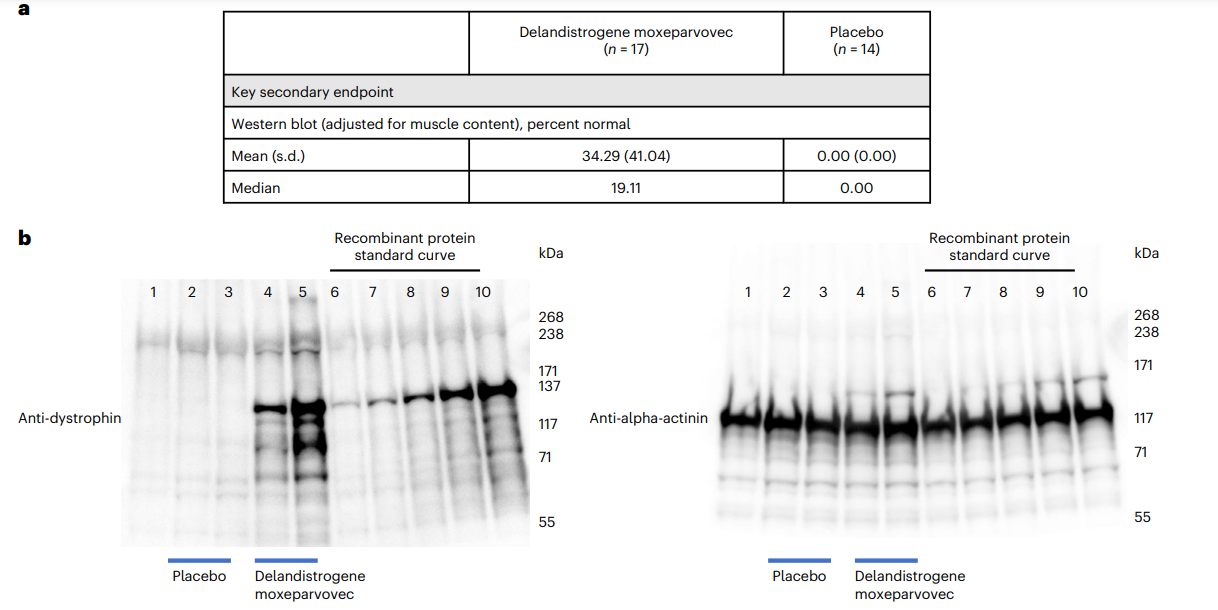
The primary objective of the study was to measure improvement in motor function using the North Star Ambulatory Assessment (NSAA) after 52 weeks. Although the therapy group showed slight improvement compared to the placebo, the difference was not statistically significant. Secondary outcomes like micro-dystrophin expression and timed motor tests (e.g., Time to Rise and 10-meter Walk/Run) showed some favorable results for the treatment group.
Safety data indicated that most side effects were mild to moderate, with no deaths or life-threatening events. Seven patients experienced serious adverse events related to the treatment, but these were resolved. Overall, the therapy was well tolerated, but its efficacy remains uncertain at this stage of the trial.

این مقاله نتایج مطالعه فاز ۳ کارآزمایی بالینی (EMBARK) را که به ارزیابی اثربخشی و ایمنی یک ژندرمانی به نام delandistrogene moxeparvovec در بیماران مبتلا به دیستروفی عضلانی دوشن (DMD) پرداخته، ارائه میکند. دیستروفی عضلانی دوشن یک بیماری ارثی شدید است که به تدریج باعث ضعف عضلانی میشود و ناشی از عدم وجود پروتئین دیستروفین است. این ژندرمانی از یک وکتور ویروسی AAV برای انتقال ژن دیستروفین به سلولهای عضلانی استفاده میکند. این مطالعه روی پسران ۴ تا ۸ ساله انجام شد که به صورت تصادفی این ژندرمانی یا دارونما دریافت کردند. هدف اصلی این مطالعه اندازهگیری بهبود عملکرد حرکتی با استفاده از ارزیابی آمبولانس ستاره شمالی (NSAA) بعد از ۵۲ هفته بود. هرچند گروه دریافتکننده درمان بهبودی اندکی نسبت به گروه دارونما نشان دادند، اما این تفاوت از نظر آماری معنیدار نبود. نتایج ثانویه مانند بیان میکرو دیستروفین و آزمایشات زمانی حرکتی (مثلاً زمان بلند شدن و ۱۰ متر دویدن) نتایج مطلوبی برای گروه درمان داشت. دادههای ایمنی نشان دادند که اکثر عوارض جانبی خفیف تا متوسط بودند و هیچ مرگ یا اتفاق تهدیدکننده زندگی رخ نداد. هفت بیمار دچار عوارض جدی مرتبط با درمان شدند که این موارد نیز حل شد. به طور کلی، درمان به خوبی تحمل شد، اما اثربخشی آن هنوز در این مرحله از کارآزمایی قطعی نیست.






| Authors | Jerry R. Mendell, Francesco Muntoni, Craig M. McDonald, Eugenio M. Mercuri, Emma Ciafaloni, Hirofumi Komaki, Carmen Leon-Astudillo, Andrés Nascimento, Crystal Proud, Ulrike Schara-Schmidt, Aravindhan Veerapandiyan, Craig M. Zaidman, Maitea Guridi, Alexander P. Murphy, Carol Reid, Christoph Wandel, Damon R. Asher, Eddie Darton, Stefanie Mason, Rachael A. Potter, Teji Singh, Wenfei Zhang, Paulo Fontoura, Jacob S. Elkins, Louise R. Rodino-Klapac |
| Corresponding Author | Jerry R. Mendell (JMendell@Sarepta.com) |
| Article Title | AAV Gene Therapy for Duchenne Muscular Dystrophy: The EMBARK Phase 3 Randomized Trial |
| Publication Date | Accepted: 17 September 2024 |
| Journal Name | Nature Medicine |
| Journal Ranking | Q1 (Quartile 1) |
| Keywords | Duchenne muscular dystrophy, gene therapy, micro-dystrophin, clinical trial, delandistrogene moxeparvovec |
| Methods Used | Randomized, double-blind, placebo-controlled trial; North Star Ambulatory Assessment, Western blot for micro-dystrophin |
| DOI | https://doi.org/10.1038/s41591-024-03304-z |