

Summary
Duchenne muscular dystrophy (DMD) is a severe, X-linked disorder characterized by progressive muscle wasting due to mutations in the dystrophin gene. Exon-skipping therapy, utilizing antisense oligonucleotides (AOs), aims to bypass mutated exons, restoring the reading frame and enabling the production of functional dystrophin, thereby converting DMD to a milder Becker muscular dystrophy (BMD) phenotype.
1. Introduction
- DMD is caused by mutations in the dystrophin gene, leading to the absence of dystrophin protein and resulting in muscle degeneration and necrosis.
- Clinical symptoms include early-onset muscle weakness, loss of ambulation by early teens, and premature death due to respiratory or cardiac failure.
- The dystrophin gene is located on the X chromosome and comprises 79 exons, with hotspot mutations often occurring between exons 3–8 and 45–55.
2. Exon-Skipping Therapy
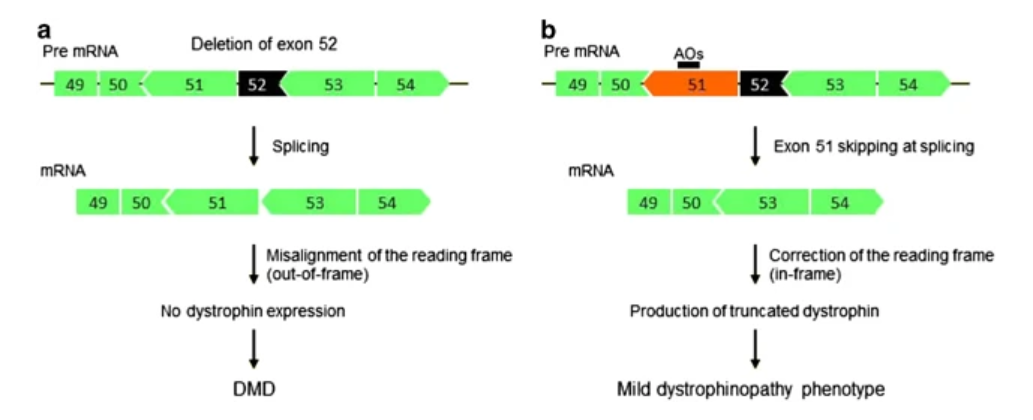
- AOs are designed to bind specific sequences of dystrophin pre-mRNA, leading to the exclusion of targeted exons during mRNA splicing.
- Effective AOs include:
- 2′-O-methyl-phosphorothioate (2′OMeAO)
- Phosphorodiamidate morpholino oligomers (PMOs)
- Exon skipping corrects the reading frame, producing a shorter but functional dystrophin protein akin to that seen in BMD.
3. Clinical Trials and FDA-Approved Drugs
- Clinical trials with:
- Drisapersen (2′OMeAO)
- Eteplirsen (PMO) – FDA approved
- NS-065/NCNP-01 (Exon 53 skipping)
- SRP-4045 (Exon 45 skipping)
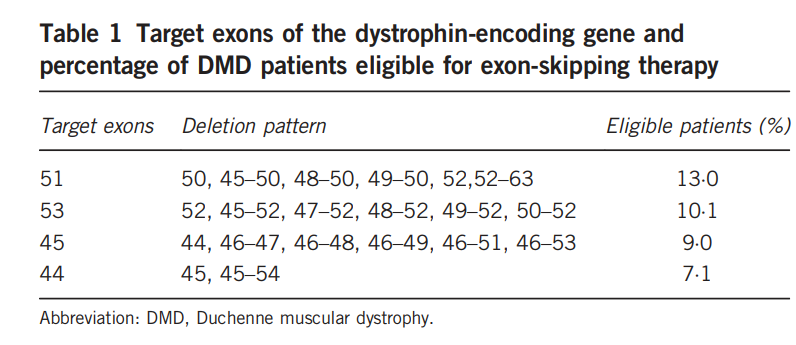
- Exon-skipping targeting exon 51 can treat ~13% of DMD patients, while exon 53 skipping can treat ~10%.
4. Challenges and Future Prospects
- Short half-life of PMOs requiring frequent administration.
- Variable efficiency across tissues, with lower dystrophin induction in heart muscle.
- Potential for multi-exon skipping (e.g., exons 45–55) to treat ~60% of DMD patients.
Conclusion
Exon-skipping therapy represents a promising genetic approach to treat DMD. However, advancements in AO chemistry, improved delivery methods, and thorough clinical evaluations based on large-cohort studies are essential to ensure its efficacy and success.

| Published | 1/1/2017 |
| Address | doi:10.1038/jhg.2017.57 |
| Authors | Akinori Nakamura |











