چشمانداز نوین در درمان دیستروفی عضلانی دوشن: ژندرمانی مبتنی بر AAV
مقدمه
دیستروفی عضلانی دوشن (DMD) یکی از شدیدترین بیماریهای ژنتیکی عضلانی است که ناشی از جهش در ژن بسیار بزرگ دیستروفین (با اندازه بیش از ۲.۲ مگاباز) میباشد. از زمان شناسایی این ژن، تلاشهای چشمگیری برای درمان این بیماری از طریق ژندرمانی صورت گرفته است. مقاله حاضر مروری جامع بر پیشرفتهای حاصل در استفاده از ناقلهای ویروسی AAV برای درمان DMD ارائه میدهد.
چالشهای اصلی در ژندرمانی DMD
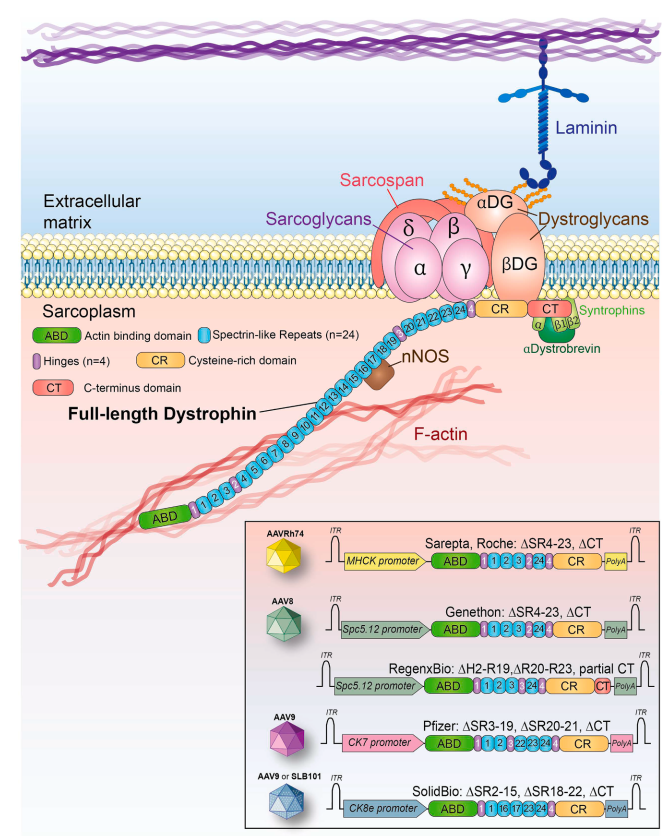
اندازه بزرگ ژن دیستروفین: امکان انتقال کل ژن با ناقلهای ویروسی مانند AAV وجود ندارد.
درگیری گسترده عضلات: درمان نیازمند انتقال ژن به بیش از ۶۵۰ عضله اسکلتی و همچنین قلب است.
پاسخ ایمنی: واکنشهای ایمنی نسبت به پروتئین دیستروفین یا کپسید ویروس میتواند موفقیت درمان را محدود کند.
استفاده از نسخههای کوتاهشده ژن
مطالعات روی بیماران مبتلا به فرم خفیفتر بیماری (Becker) نشان داد که نسخههای کوچکتری از دیستروفین (مانند مینیدیستروفین و میکرودیستروفین) نیز عملکرد نسبی دارند. این امر موجب توسعه نسخههای مصنوعی بهینهسازیشده از ژن شد که در قالب AAV قابل انتقال هستند.
موفقیتها و محدودیتهای بالینی
شش شرکت بیوتکنولوژی تاکنون ژندرمانی مبتنی بر AAV/μDys را وارد فازهای مختلف کارآزمایی کردهاند. تنها یکی از این روشها (محصول Sarepta) در سال 2024 تأیید FDA را دریافت کرد. نتایج بالینی تاکنون نشاندهندهی بهبود نسبی در علائم بیماران بوده اما اثربخشی کامل هنوز حاصل نشده است. همچنین برخی عوارض جدی از جمله افزایش آنزیمهای کبدی، مشکلات ایمنی و حتی چند مورد مرگ نیز گزارش شدهاند.
نوآوری در طراحی ناقلهای ویروسی
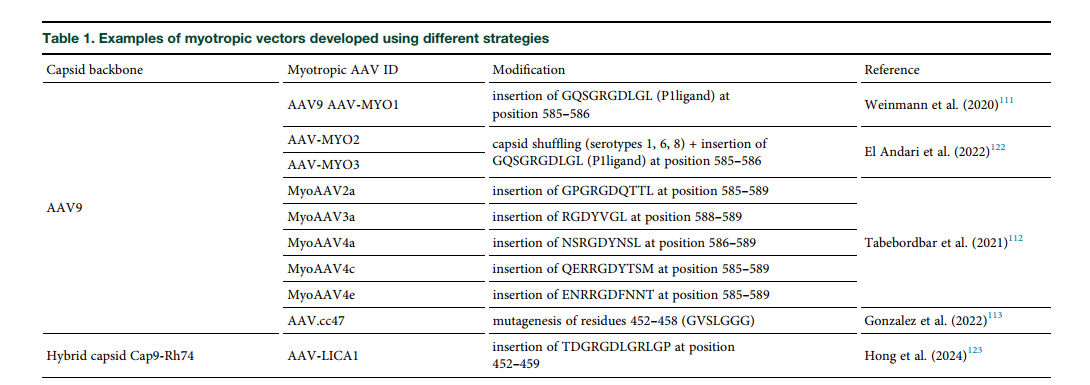
تحقیقات اخیر بر روی توسعهی کپسیدهای ویروسی خاص (myotropic AAVs) متمرکز شده است که توانایی هدفگیری اختصاصی عضله را در دوزهای کمتر دارند و عوارض کبدی را کاهش میدهند. نخستین نتایج این ناقلها در انسان امیدوارکننده بوده است.
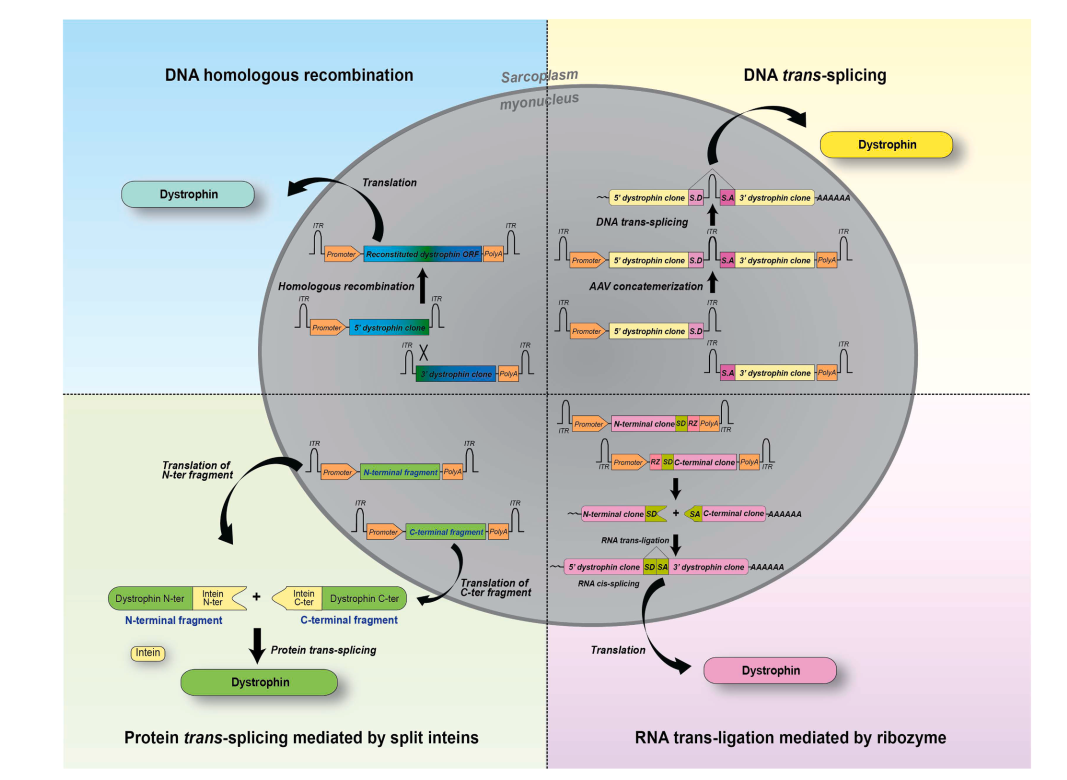
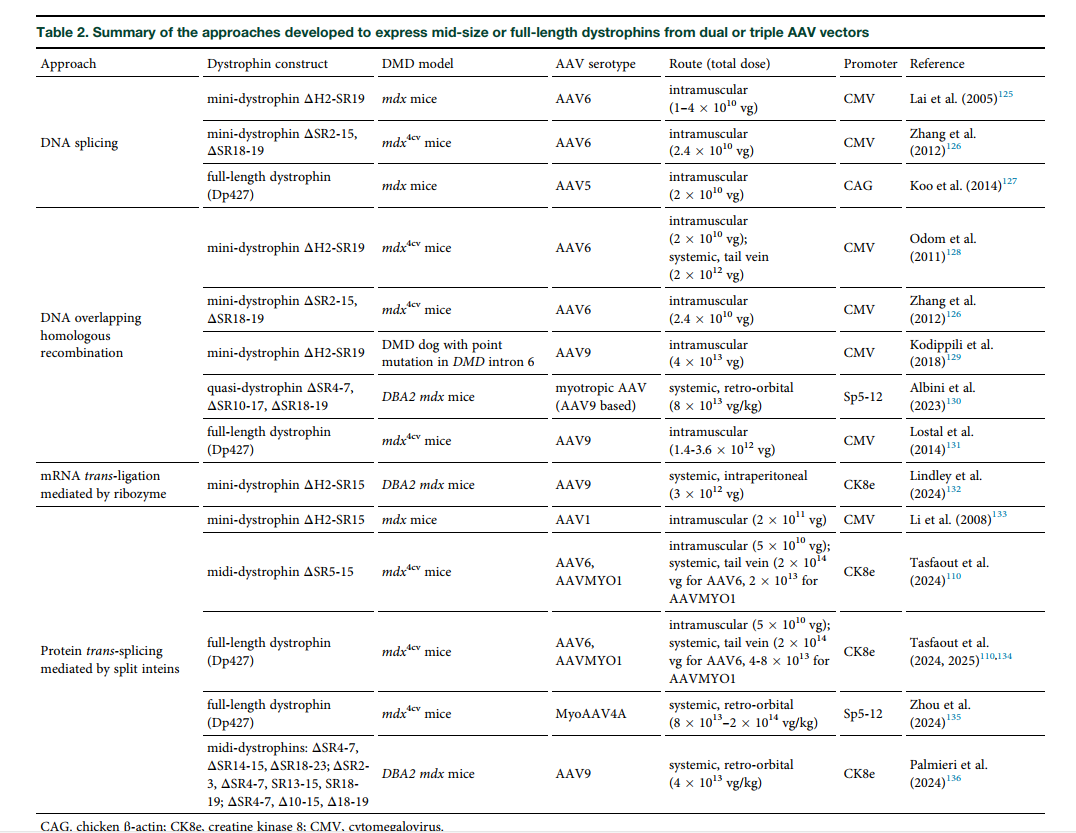
عبور از محدودیت ظرفیت AAV
برای انتقال نسخههای بزرگتر دیستروفین، استراتژیهای جدیدی مانند:
- ✅ استفاده از ناقلهای دوگانه یا سهگانه برای بازسازی ژن کامل
- ✅ اتصال قطعات پروتئینی با استفاده از اینتئینها (inteins)
- ✅ روشهای RNA محور نظیر ribozyme
توسعه یافتهاند. با این حال، کارایی این روشها در مدلهای حیوانی محدود باقی مانده و نیاز به بهینهسازی دارد.
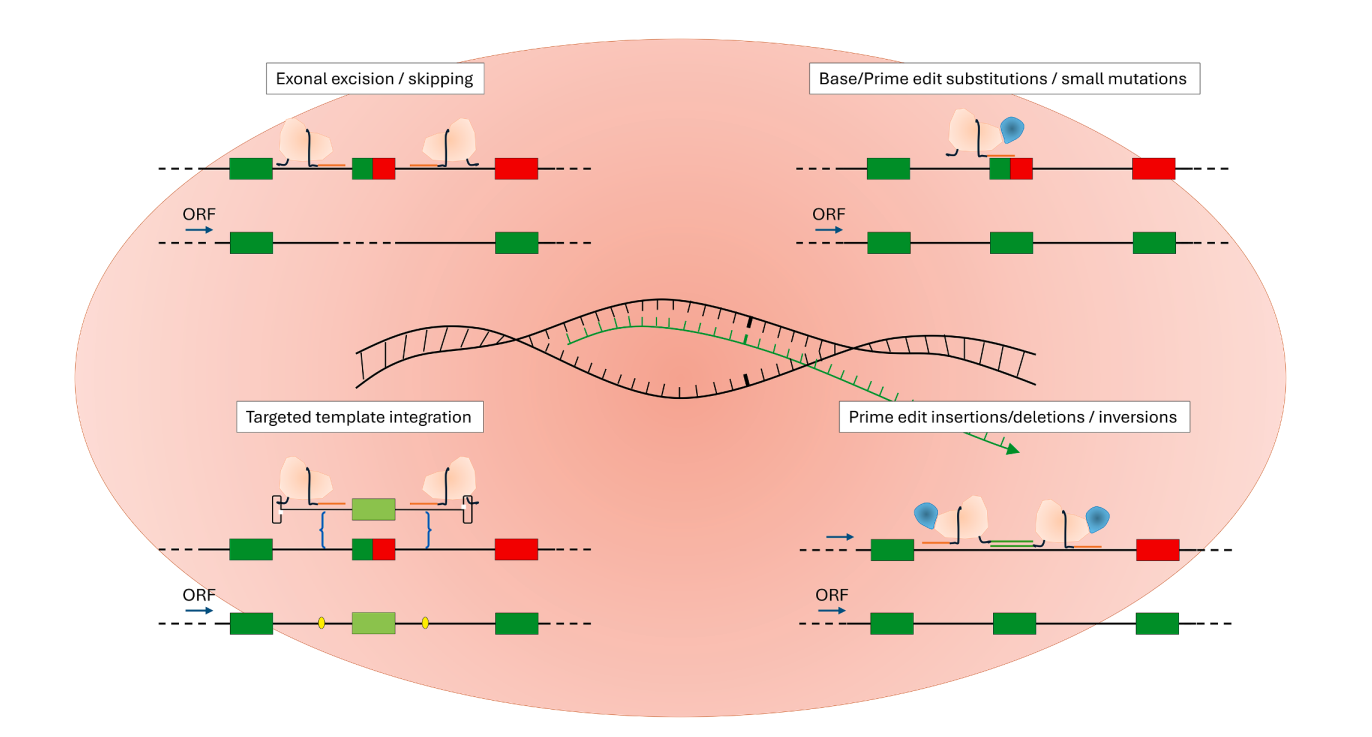
چشمانداز ویرایش ژنوم
ویرایش ژن با سیستم CRISPR-Cas9 توانسته است در مدلهای حیوانی بیان طبیعی دیستروفین را بازگرداند. روشهای دقیقتری مانند Base Editing و Prime Editing نیز در حال بررسی هستند. با وجود چالشهایی مانند ایمنی و کارایی، این رویکردها امیدبخشترین مسیر برای درمان دائمی DMD تلقی میشوند.
نتیجهگیری
گرچه اولین محصول ژندرمانی DMD به بازار راه یافته است، مسیر درمان کامل این بیماری همچنان در حال توسعه است. آیندهی درمان DMD متکی بر بهبود ناقلها، طراحیهای ژنی نوین، و تکنولوژیهای ویرایش ژن خواهد بود. پیشرفتهای سریع این حوزه نویدبخش درمان مؤثرتر و پایدارتر در آیندهای نزدیک است.



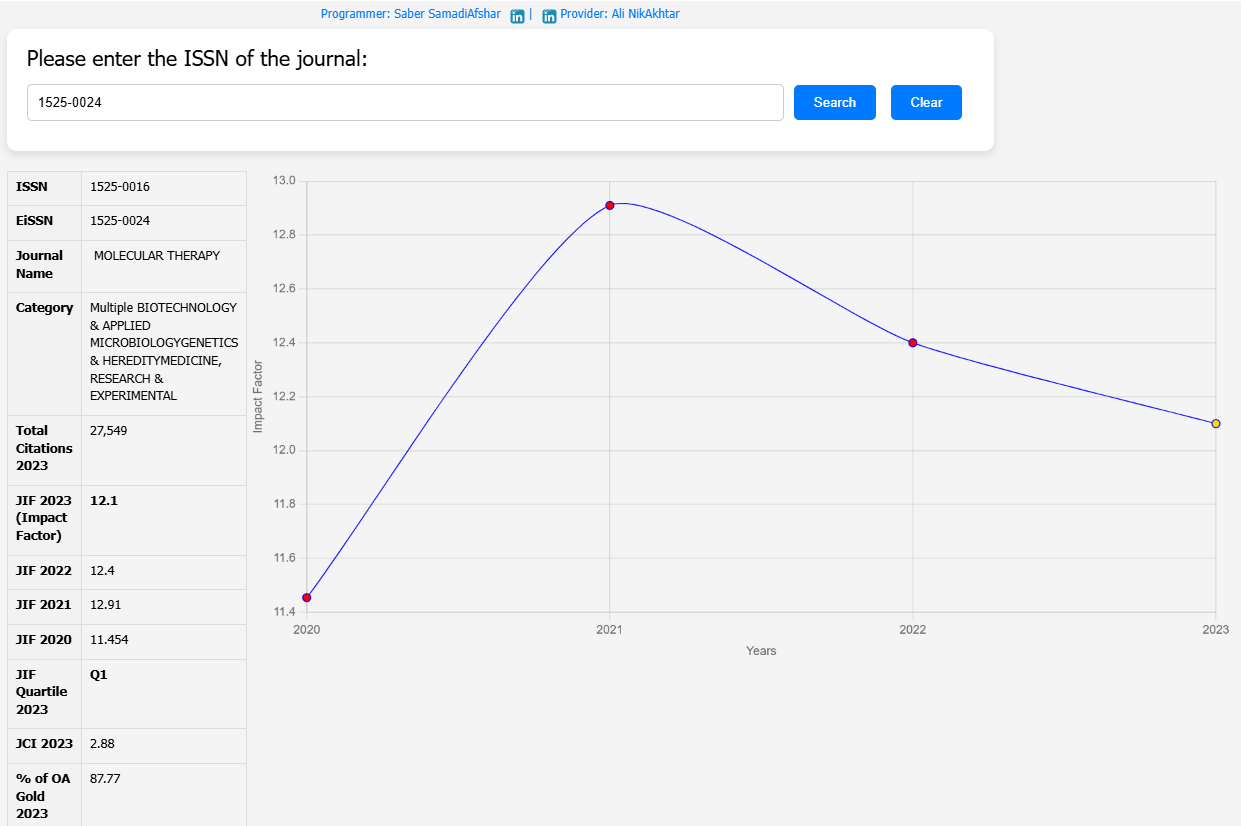
| Field | Details |
|---|---|
| Title | The road toward AAV-mediated gene therapy of Duchenne muscular dystrophy |
| Authors | Niclas E. Bengtsson, Hichem Tasfaout, Jeffrey S. Chamberlain |
| Corresponding Author | Niclas E. Bengtsson (niclasb@uw.edu) Hichem Tasfaout (tasfaout@uw.edu) Jeffrey S. Chamberlain (jsc5@uw.edu) |
| Publication Date | May 2025 |
| Journal | Molecular Therapy, Vol. 33, No. 5 |
| Keywords | Duchenne muscular dystrophy, gene therapy, AAV vectors, micro-dystrophin, CRISPR-Cas9, myotropic AAV, muscle-targeted delivery |
| Methods Used | Review of preclinical and clinical studies including: – AAV vector development – micro/midi/full-length dystrophin delivery – split-intein protein trans-splicing – gene editing (CRISPR, base/prime editing) – dual/triple vector strategies |
| Study Type | Review Article |
| DOI | 10.1016/j.ymthe.2025.03.065 |