

Abstract
This study investigates the transcriptional dysregulation of autophagy in the skeletal muscles of mdx mice, a model for Duchenne muscular dystrophy (DMD). The focus is on the roles of FoxO transcription factors (FoxO1 and FoxO3a) and transcription factor EB (TFEB). The findings reveal significant downregulation of these genes due to phosphorylation-mediated suppression.
Key Findings
- Global Downregulation of Autophagy-Related Genes: Microarray and qPCR analyses revealed reduced expression of genes involved in:
- Autophagosome formation
- Autophagosome-lysosome fusion
- Mitophagy
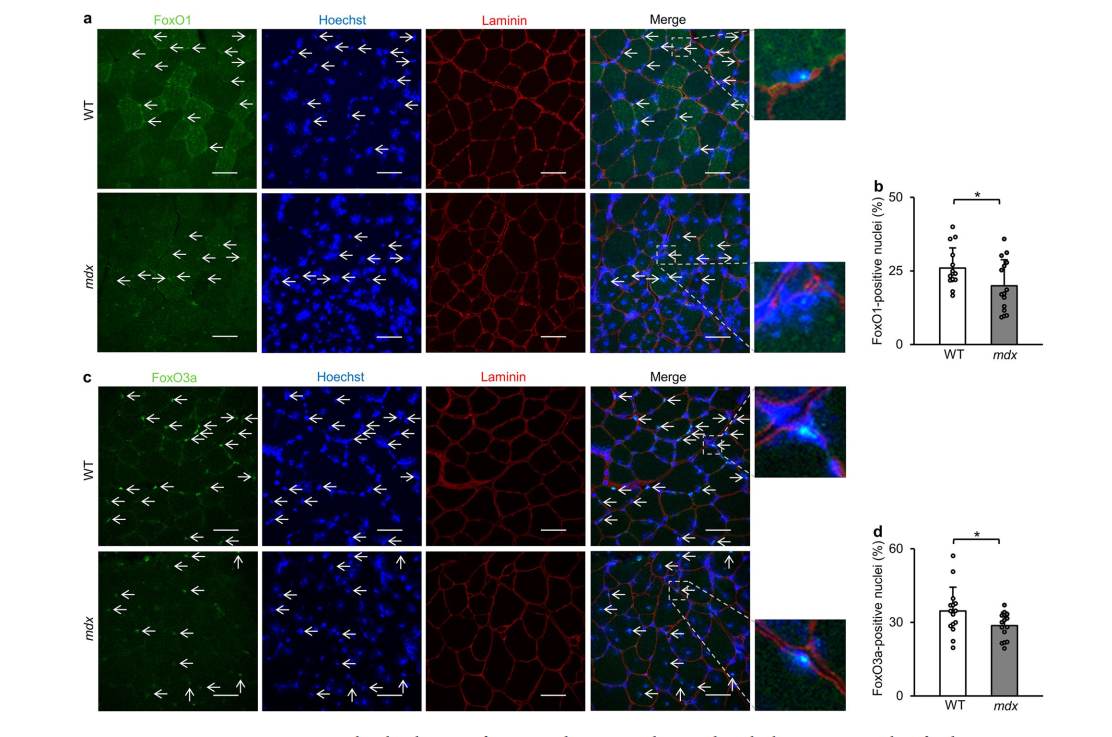
- FoxO1 and FoxO3a Dysregulation: Decreased nuclear localization and increased phosphorylation of FoxO1 (Thr24) and FoxO3a (Thr32), suggesting impaired transcriptional activity due to Akt-mediated phosphorylation.
- TFEB Dysregulation: Reduced nuclear localization and increased phosphorylation of TFEB (Ser122) in mdx mice, indicating mTORC1-mediated suppression of TFEB activity.
- Accumulation of Autophagic Markers: Elevated levels of LC3-II and p62 proteins, indicating impaired autophagosome degradation.
- Resveratrol as a Potential Therapeutic Agent: Resveratrol, an activator of SIRT1, was shown to restore autophagy-related gene expression, decrease phosphorylation of TFEB, and ameliorate muscle pathology.
Mechanistic Insights
- Akt-mTORC1 Pathway: Increased Akt-mTORC1 signaling in dystrophic muscles leads to:
- Increased phosphorylation of FoxOs and TFEB
- Nuclear export and suppression of autophagy-related genes
- FoxOs and TFEB: Crucial for maintaining autophagic activity. Their suppression leads to:
- Impaired degradation of damaged proteins and organelles
- Muscle degeneration and atrophy
Clinical Implications
Targeting the transcriptional regulation of autophagy could lead to novel therapeutic strategies for DMD. Correcting autophagy dysregulation may:
- Reduce muscle degeneration
- Improve clinical outcomes for DMD patients
Conclusion
This study provides compelling evidence that transcriptional dysregulation of autophagy, mediated by phosphorylation-dependent suppression of FoxOs and TFEB, plays a critical role in the pathophysiology of DMD. Therapeutic interventions restoring the activity of these transcription factors hold promise for improving muscle health in DMD.
| Published | 16/01/2024 |
| Address | DOI: 10.1038/s41598-024-51746-9 |
| Authors | Ryuta Nakashima 1, Ryusuke Hosoda 1, Yuki Tatekoshi 1, Naotoshi Iwahara 1 2, Yukika Saga 1, Atsushi Kuno 3 |











