

Overview
Duchenne muscular dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness due to the absence of dystrophin. This study investigates the disruption of autophagy, a cellular process responsible for degrading damaged proteins and organelles, in the skeletal muscles of DMD patients and mdx mice, a common model for DMD.
Key Findings
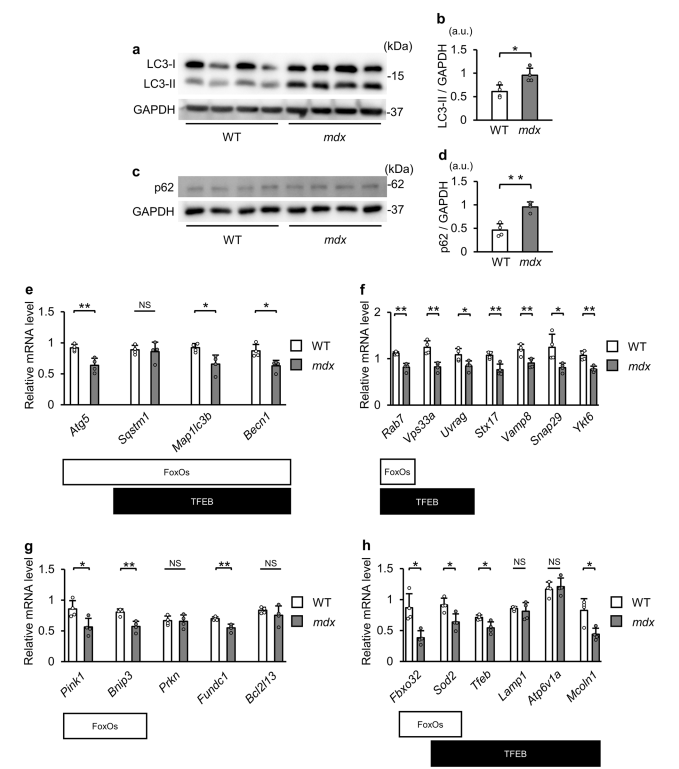
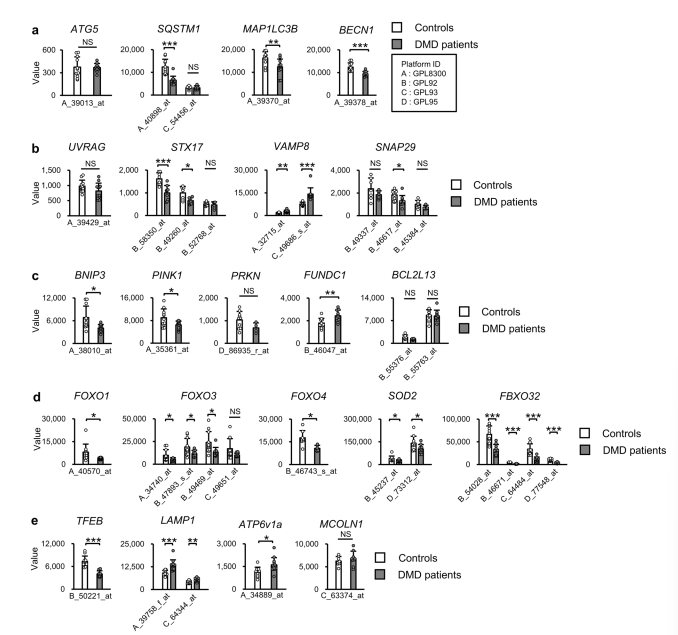
- Downregulation of Autophagy-Related Genes: Autophagy-related genes are significantly downregulated in dystrophin-deficient muscles.
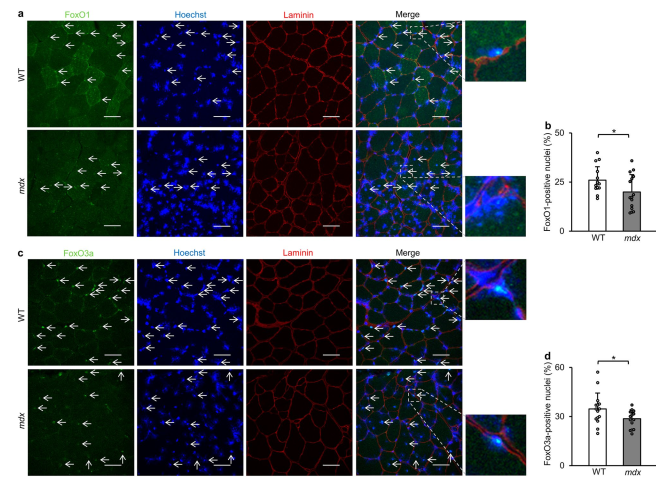
- Phosphorylation of Key Transcription Factors: FoxO and TFEB are phosphorylated, preventing their nuclear entry and reducing autophagy-related gene expression.
- Impaired Autophagic Process: The failure in autophagosome-lysosome fusion leads to the accumulation of autophagosomes.
Potential Therapeutic Approaches
- SIRT1 Activation with Resveratrol: Resveratrol reduces the phosphorylation of FoxO and TFEB, restoring autophagic activity and improving muscle function in mdx mice.
- Transcriptional Regulation Strategies: Targeting transcriptional dysregulation of autophagy-related genes could be a promising approach for mitigating muscle degeneration in DMD.
Conclusion
This study highlights the importance of correcting the transcriptional suppression of autophagy-related genes in DMD to restore proper autophagic function. Future research should explore additional transcription factors and epigenetic mechanisms involved in this process to develop more effective treatments for DMD.
| Published | 1/16/2024 |
| Address | https://doi.org/10.1038/s41598-024-51746-9 |
| Authors | Ryuta Nakashima1, Ryusuke Hosoda1, Yuki Tatekoshi1, Naotoshi Iwahara1,2, Yukika Saga1 & Atsushi Kuno |











