

Overview
Duchenne Muscular Dystrophy (DMD) is a rare genetic disorder characterized by progressive muscle degeneration, often leading to severe cardiac complications. This systematic review analyzed and graded existing literature on predictors of cardiac disease in DMD patients.
Research Methodology
The study reviewed 33 publications published between 2000 and 2022, involving 9,232 patients. Researchers focused on:
- Pharmacological treatments: Evaluating drugs that influence cardiac health.
- Genetic mutations: Assessing their role in disease progression.
- Non-pharmacological interventions: Investigating additional factors influencing cardiac function.
- Utilizing the GRADE framework to assess the certainty of evidence.
Key Findings
1. Pharmacological Treatments Improve Cardiac Outcomes
- ACE inhibitors: Shown to improve cardiac function by preserving left ventricular ejection fraction (LVEF).
- β-blockers: Associated with reduced cardiomyopathy risk.
- Mineralocorticoid receptor antagonists: Help maintain cardiac health.
- Glucocorticoids (Deflazacort): High-quality evidence supports its role in preserving LVEF and delaying cardiac dysfunction.
2. Genetic Factors Influence Cardiac Risk
- Mutations in exons 51 and 52: Linked to lower risk of severe cardiac issues.
- Exon skipping therapies: Help extend cardiac function.
- Genetic modifiers (LTBP4, ACTN3): Potentially affect cardiac outcomes, though evidence is less certain.
3. Mechanical Ventilation and Cardiac Function
- Mechanical ventilation: Shows a modest correlation with improved cardiac function.
- More research is needed on non-pharmacological interventions.
Future Directions
While there is moderate to high-quality evidence supporting cardiac medications in managing heart disease in DMD patients, the research highlights several areas that need further exploration:
- Improved study designs: Reducing data inconsistencies in clinical trials.
- Exploring non-pharmacological treatments: Understanding their impact on cardiac health.
- Refining therapy initiation criteria: Identifying the optimal timing for starting cardiac interventions.
Conclusion
This review underscores the importance of early cardiac management in DMD patients. While pharmacological therapies offer significant benefits, further research is needed to explore genetic factors, non-drug interventions, and long-term cardiac outcomes to improve patient care.
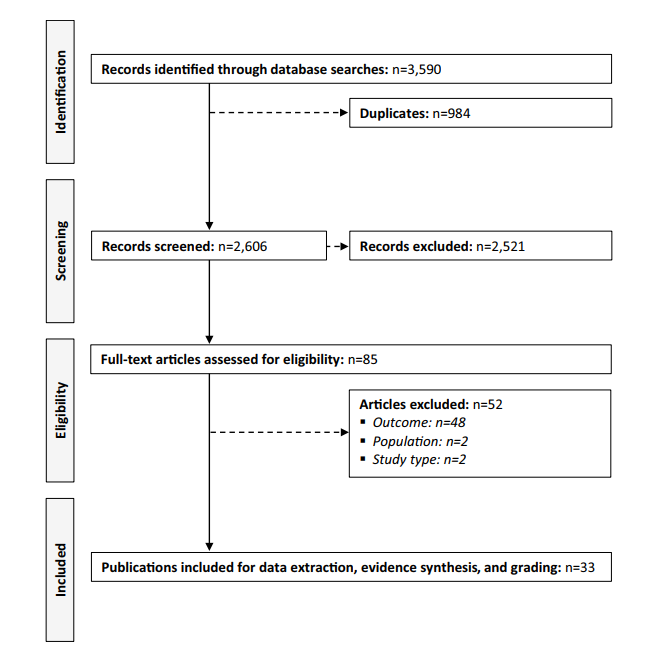
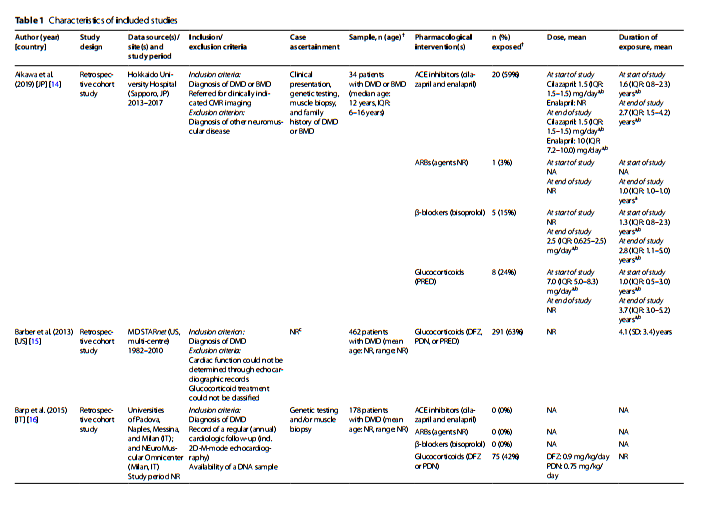






| Published | 28/09/2024 |
| Address | https://doi.org/10.1186/s13023-024-03372-x |
| Authors | Erik Landfeldt1* , Alberto Alemán2,3, Sophia Abner4, Rongrong Zhang5 , Christian Werner6 , Ioannis Tomazos7 , Hanns Lochmüller2,3,8, Ros M. Quinlivan9 and Karim Wahbi |











