Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder caused by mutations in the dystrophin gene on the X chromosome. These mutations disrupt the production of the dystrophin protein, crucial for muscle stability and function. This review focuses on exon-skipping therapy, which uses genetic techniques to restore partial dystrophin production, converting the severe DMD phenotype to a milder Becker Muscular Dystrophy (BMD)-like condition.
Mechanism of Exon-Skipping Therapy
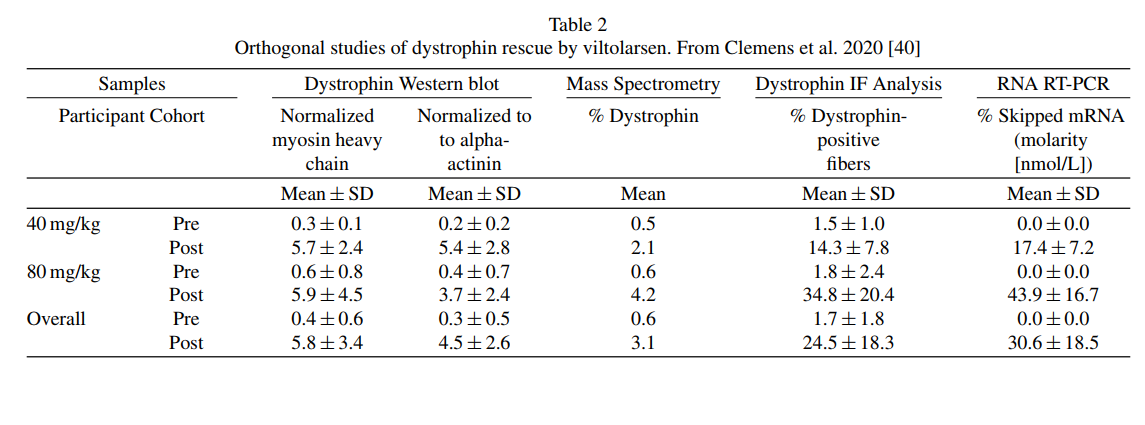
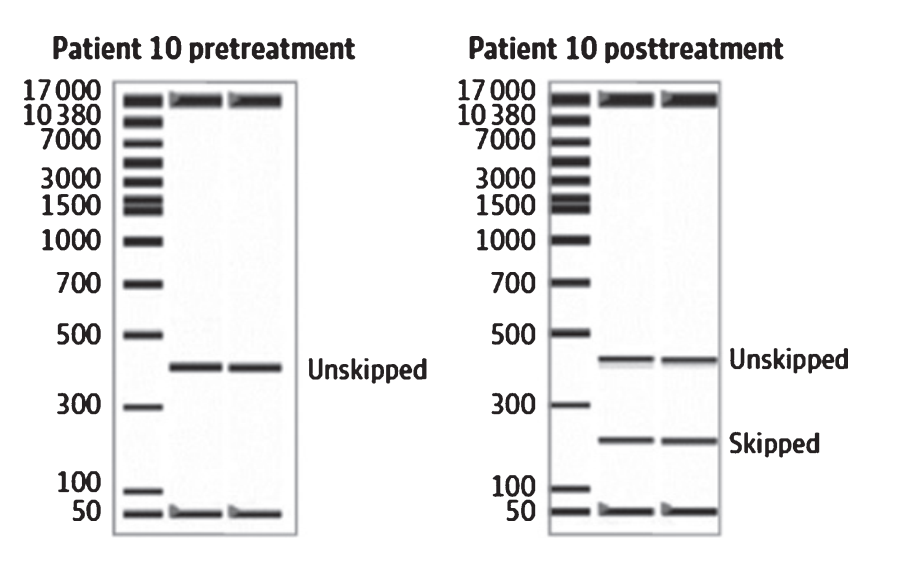
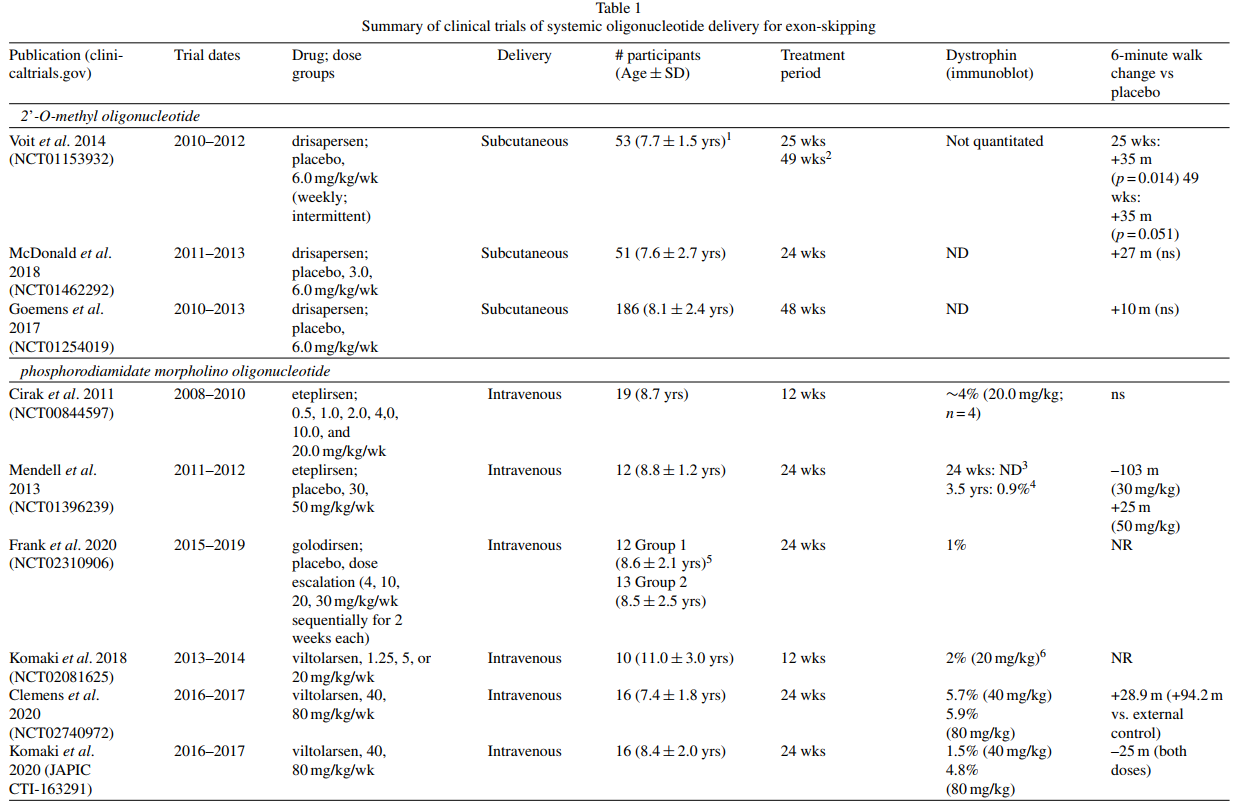
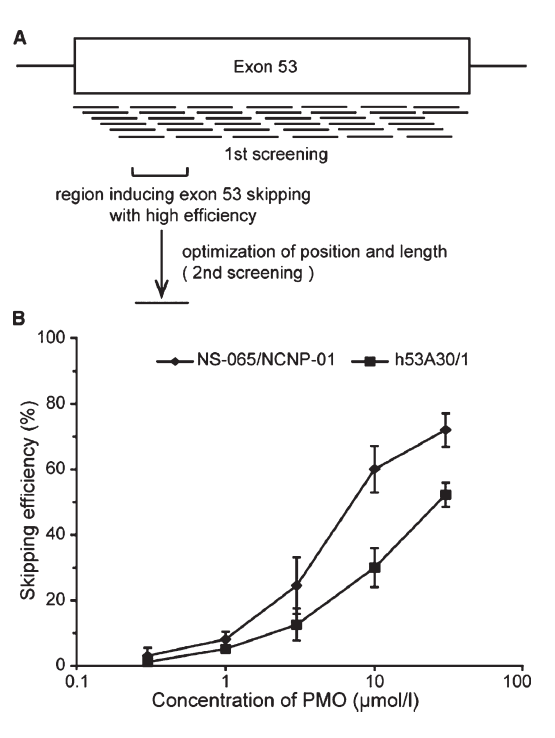
Exon-skipping therapy involves antisense oligonucleotides (AONs) that target specific exons in the dystrophin pre-mRNA. For instance, skipping exon 51 can restore the reading frame for many mutations. This approach enables the production of truncated but functional dystrophin proteins. Drugs like eteplirsen and viltolarsen have shown promise, with clinical trials demonstrating increases in dystrophin levels in patient muscles.
Limitations and Challenges
Despite progress, exon-skipping therapies have limitations. They generally lead to low levels of dystrophin restoration and variable clinical outcomes among patients. Factors like the mutation type, muscle condition, and drug delivery efficiency significantly influence treatment success. Higher doses and optimized delivery methods are areas of ongoing research to enhance efficacy.
Complementary Therapeutic Approaches
Gene therapy and stop codon read-through represent complementary strategies to treat DMD. Gene therapy introduces micro-dystrophins, smaller but functional protein versions, using viral vectors. Stop codon read-through drugs, such as ataluren, aim to bypass premature stop codons to allow dystrophin synthesis. Both approaches show potential but face challenges like immune responses and delivery barriers.
Conclusion
Early intervention in younger patients is critical for better outcomes, as advanced muscle degeneration limits the therapy’s effectiveness. Combining exon-skipping with anti-inflammatory treatments and muscle regeneration strategies may further improve patient quality of life. However, achieving consistent dystrophin levels across diverse patient populations remains a challenge.




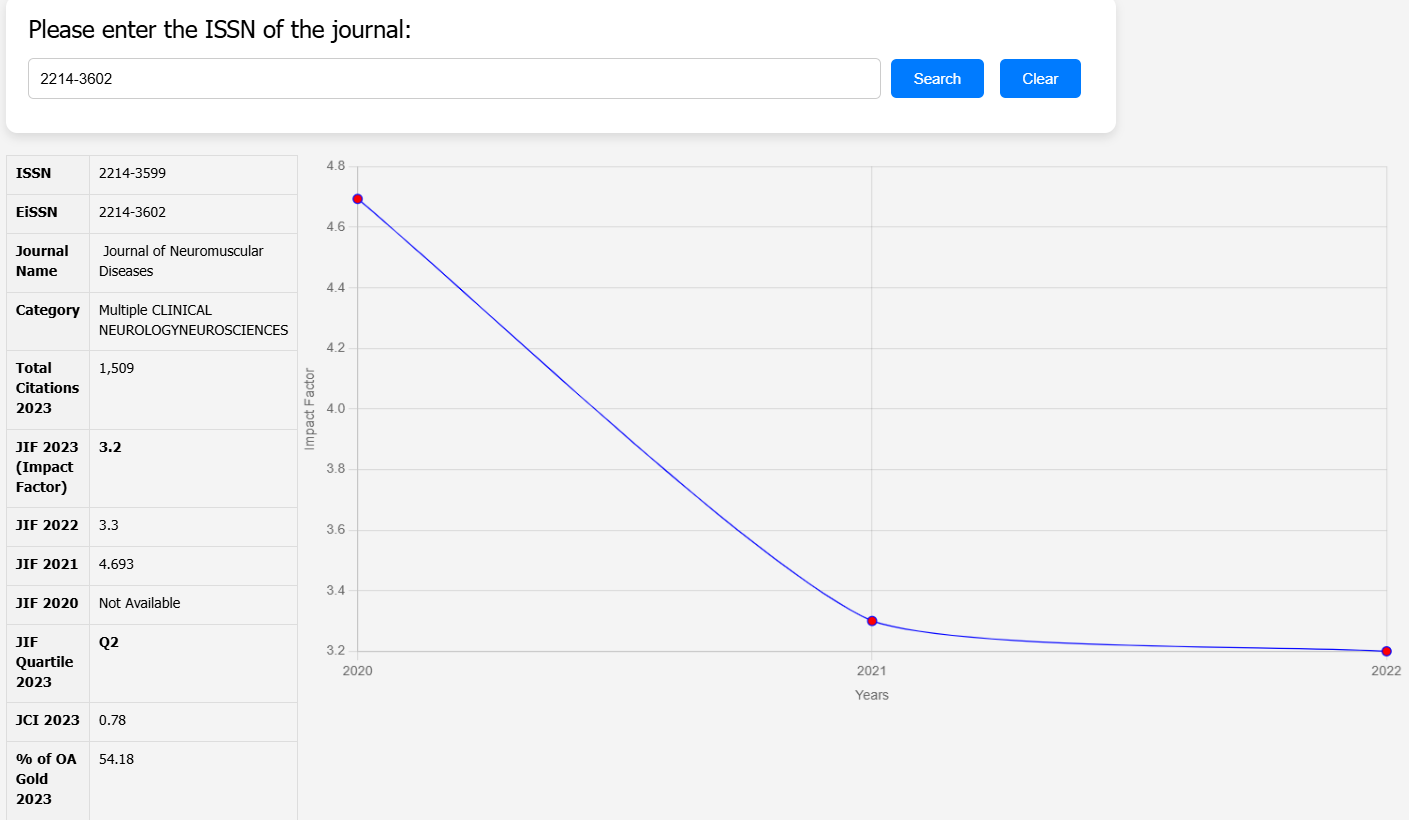
| Category | Details |
| Authors | Shin’ichi Takeda, Paula R. Clemens, Eric P. Hoffman |
| Corresponding Author | Eric P. Hoffman |
| Article Title | Exon-Skipping in Duchenne Muscular Dystrophy |
| Publication Date | June 24, 2021 (Pre-press) |
| Journal Name | Journal of Neuromuscular Diseases |
| Keywords | Exon skipping, Duchenne Muscular Dystrophy, Gene Therapy, Dystrophin, Oligonucleotide Therapy, Muscle Disorders |
| Methods Used | Antisense oligonucleotides (AONs), gene therapy, stop codon read-through, exon deletion (CRISPR), U7 snRNP-mediated splicing |
| DOI | 10.3233/JND-210682 |