1. Introduction
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder predominantly affecting boys, with an incidence of approximately 1 in 5,000 male births. It is caused by mutations in the DMD gene that lead to disrupted production of dystrophin, a protein essential for muscle stability. Exon skipping is a therapeutic strategy that restores the reading frame of the mutated gene to enable dystrophin production. This study focuses on applying this method to treat patients with rare single exon duplications.
2. Study Objective and Rationale
The study aimed to evaluate the effectiveness of casimersen and golodirsen, two exon-skipping therapies, in DMD patients with exon 45 or 53 duplications. Unlike common out-of-frame deletions, single exon duplications are rare but can theoretically be corrected to restore the production of full-length dystrophin. The primary goal was to assess dystrophin expression and clinical outcomes following treatment.
3. Methods and Participants
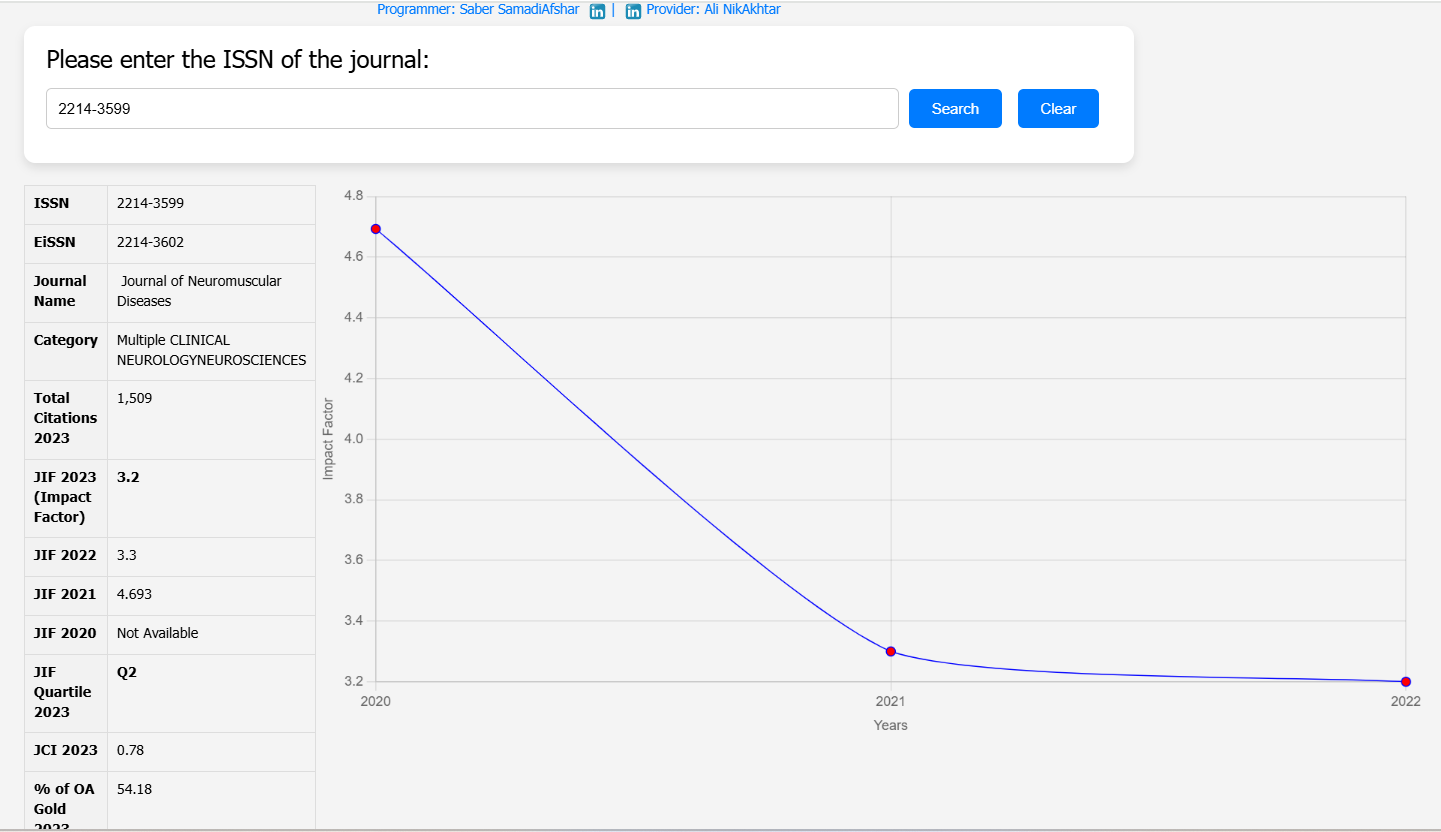
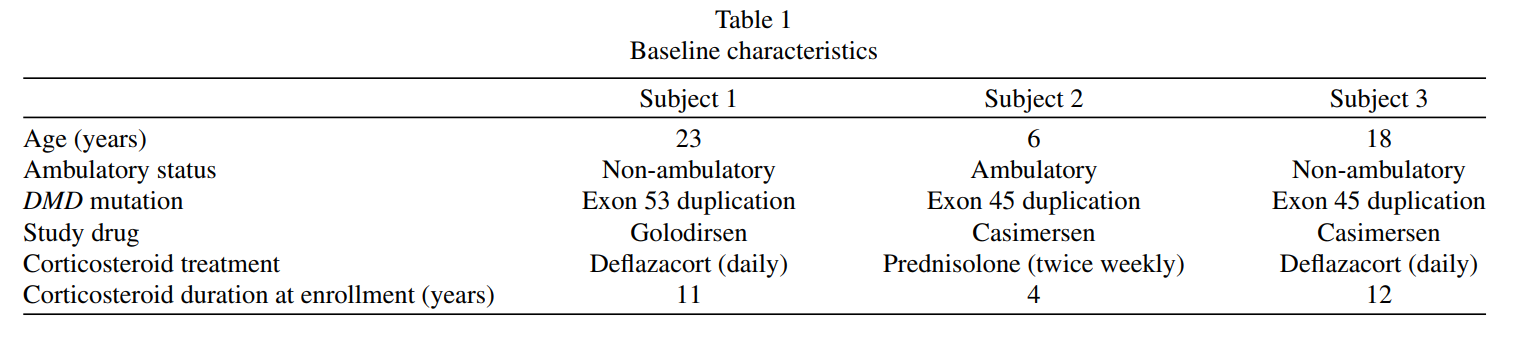
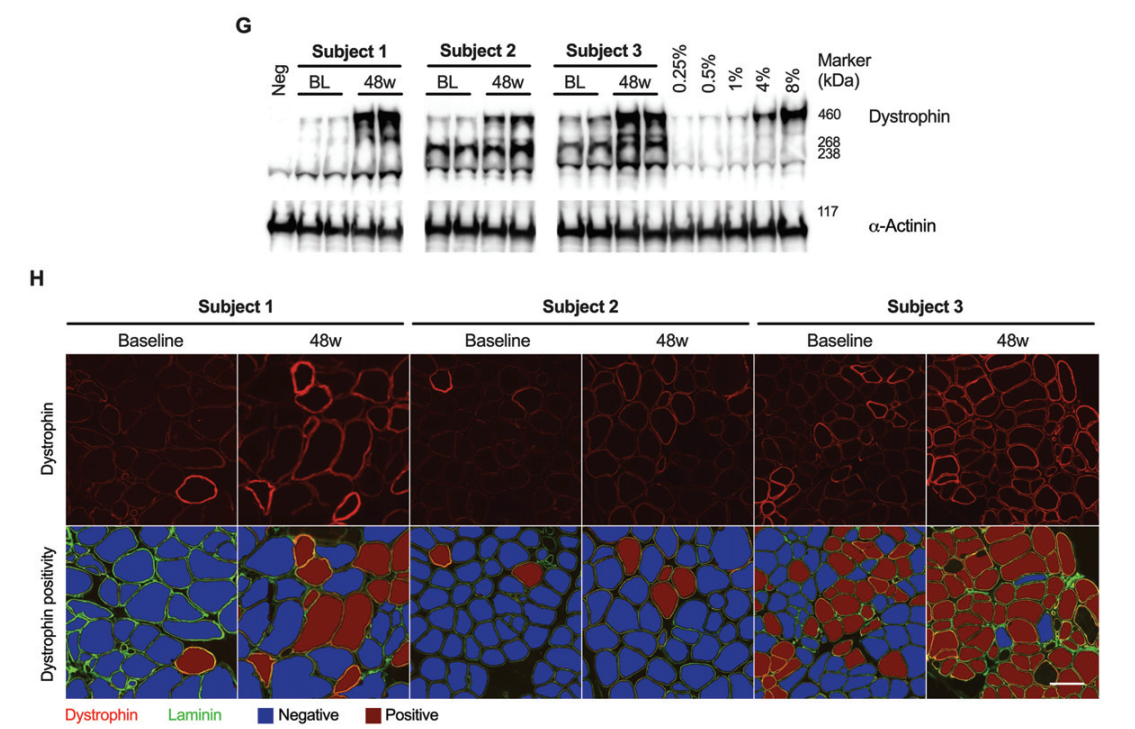
Three male participants with DMD and duplications of exon 45 or 53 were included in this open-label study. The participants were aged 6, 18, and 23 years; two were non-ambulatory, and one was ambulatory. Over 48 weeks, they received weekly intravenous doses of either casimersen or golodirsen (30 mg/kg). Muscle biopsies taken at baseline and the end of the study were analyzed using Western blot and immunofluorescence to measure dystrophin levels.
4. Key Findings
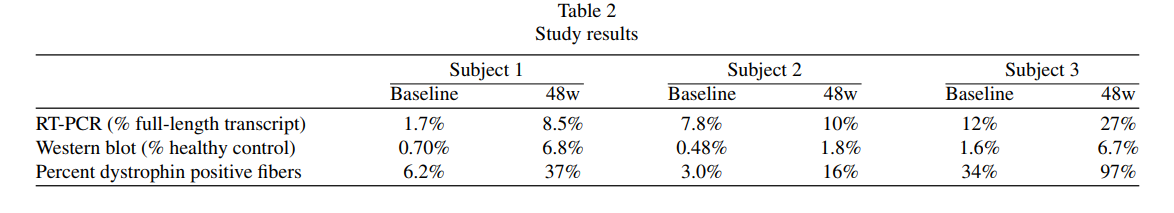
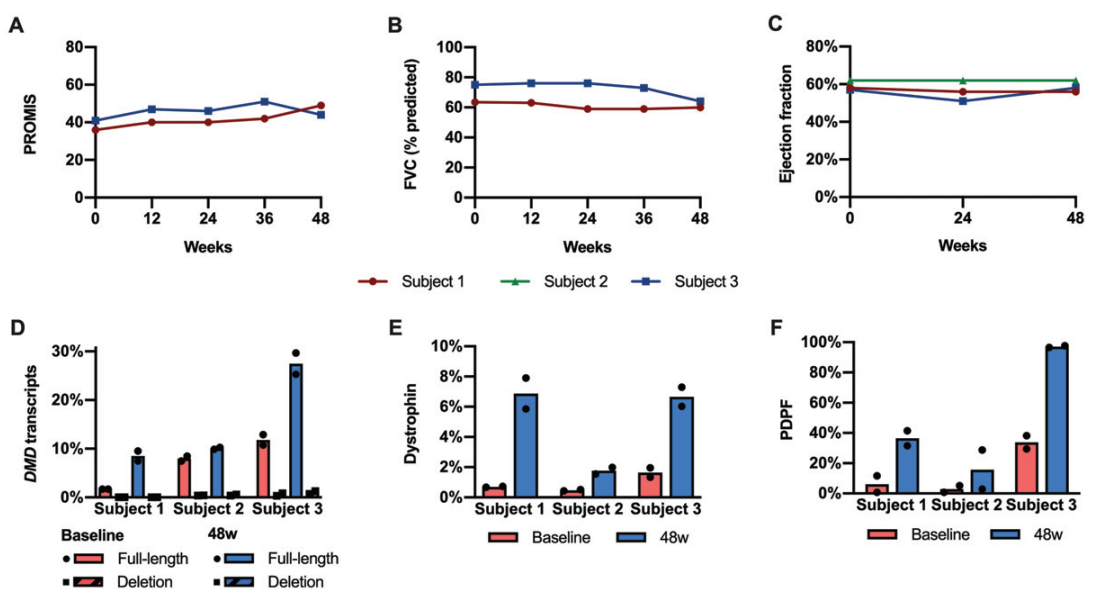
Dystrophin Expression: The average dystrophin levels increased from 0.94% of normal at baseline to 5.1% after 48 weeks. Similarly, dystrophin-positive fibers increased from 14% to 50%.
Transcript Skipping: RT-PCR analysis showed an increase in full-length DMD transcripts, validating the effectiveness of exon skipping.
Clinical Stability: Pulmonary and cardiac functions remained stable, and no major safety issues were observed during the trial.
5. Significance of Results
The findings suggest that exon-skipping therapies can significantly increase dystrophin expression in patients with rare single exon duplications. Full-length dystrophin restoration may offer superior therapeutic benefits compared to therapies producing truncated dystrophin isoforms.
6. Safety and Limitations
The study reported no serious adverse events. Mild side effects, such as headaches and rashes, were observed but manageable. However, the small sample size and short study duration limit the ability to draw conclusions about long-term safety and clinical efficacy.
7. Future Directions
The study highlights the need to develop more potent exon-skipping agents and explore combination therapies. Future trials should include larger cohorts, longer follow-up periods, and detailed assessments of functional outcomes.
8. Conclusion
Exon-skipping therapies like casimersen and golodirsen demonstrate promising potential for treating DMD patients with rare single exon duplications. This study provides initial evidence for their efficacy in restoring dystrophin, paving the way for more targeted treatments for rare genetic mutations in DMD.







| Category | Details |
| Authors | Stefan Nicolau, Jyoti Malhotra, Maryann Kaler, Pamela Magistrado Coxen, et al. |
| Corresponding Author | Kevin M. Flanigan |
| Article Title | Increase in Full-Length Dystrophin by Exon Skipping in Duchenne Muscular Dystrophy Patients with Single Exon Duplications: An Open-label Study |
| Publication Date | 30-Apr-24 |
| Journal Name | Journal of Neuromuscular Diseases |
| Keywords | DuchenneMuscularDystrophy, Dystrophin, ExonSkipping, GeneTherapy, etc. |
| Methods Used | Western blot, immunofluorescence, RT-PCR, clinical assessments |
| DOI | 10.3233/JND-230107 |