

Understanding Myotonic Dystrophy (DM): Causes, Symptoms, and Emerging Treatments
Overview
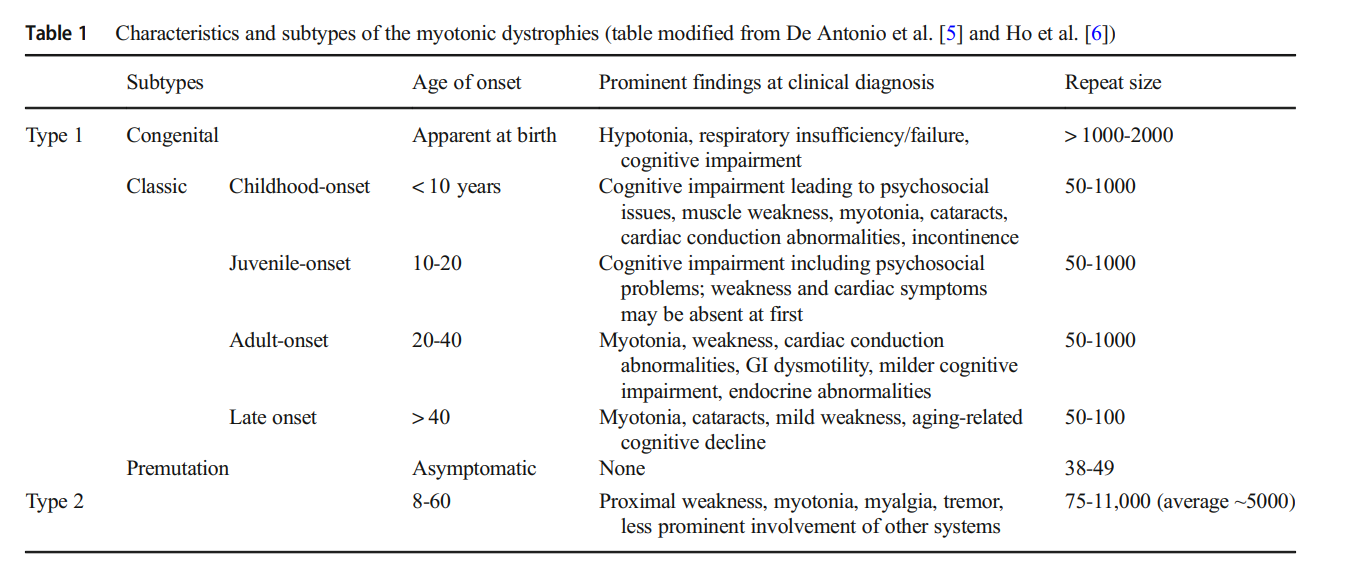
Myotonic Dystrophy (DM) is an inherited, multisystemic disorder that affects muscles and various organs. There are two primary types:
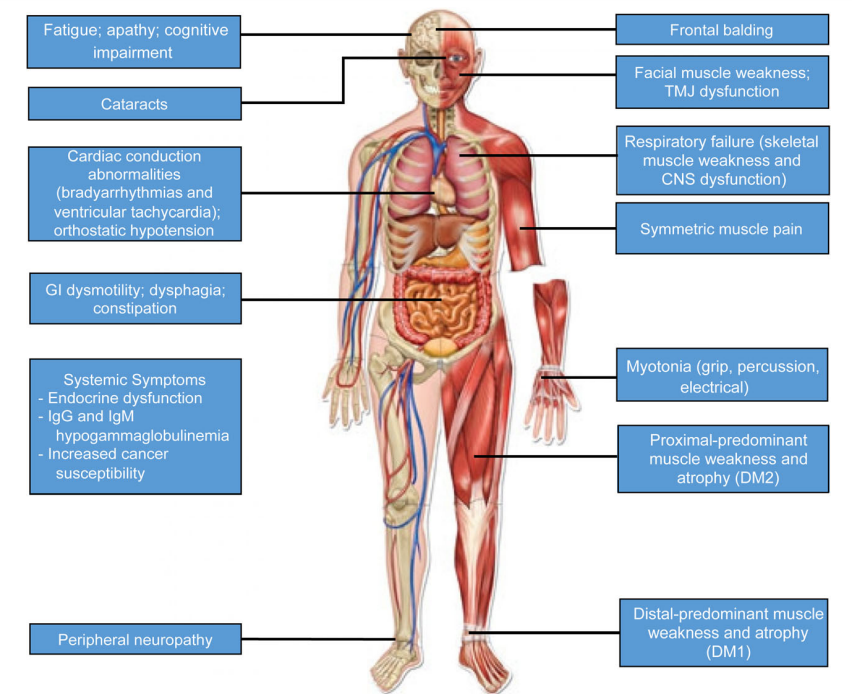
- DM1: Caused by CTG repeat expansion in the DMPK gene. It is more common and severe, leading to muscle weakness, myotonia, and systemic complications such as:
- Cardiac issues (arrhythmias).
- Respiratory complications.
- Gastrointestinal dysfunction.
- DM2: Caused by CCTG repeat expansion in the CNBP gene. It presents with less severe systemic symptoms but remains a debilitating disorder.
Pathogenesis: How Myotonic Dystrophy Affects the Body
Both DM1 and DM2 result from toxic RNA gain-of-function, where mutant RNA sequesters RNA-binding proteins, leading to faulty RNA splicing. This affects the function of multiple proteins across various tissues, causing widespread systemic effects.
Current Treatment Approaches
There are currently no disease-modifying treatments, but supportive care is available to manage symptoms.
- Cardiac complications: Managed with pacemakers or defibrillators.
- Respiratory issues: Treated with ventilation support.
- Myotonia: Treated with sodium channel blockers like mexiletine.
- Pain management: Includes physical therapy and medications.
Emerging Research and Future Therapies
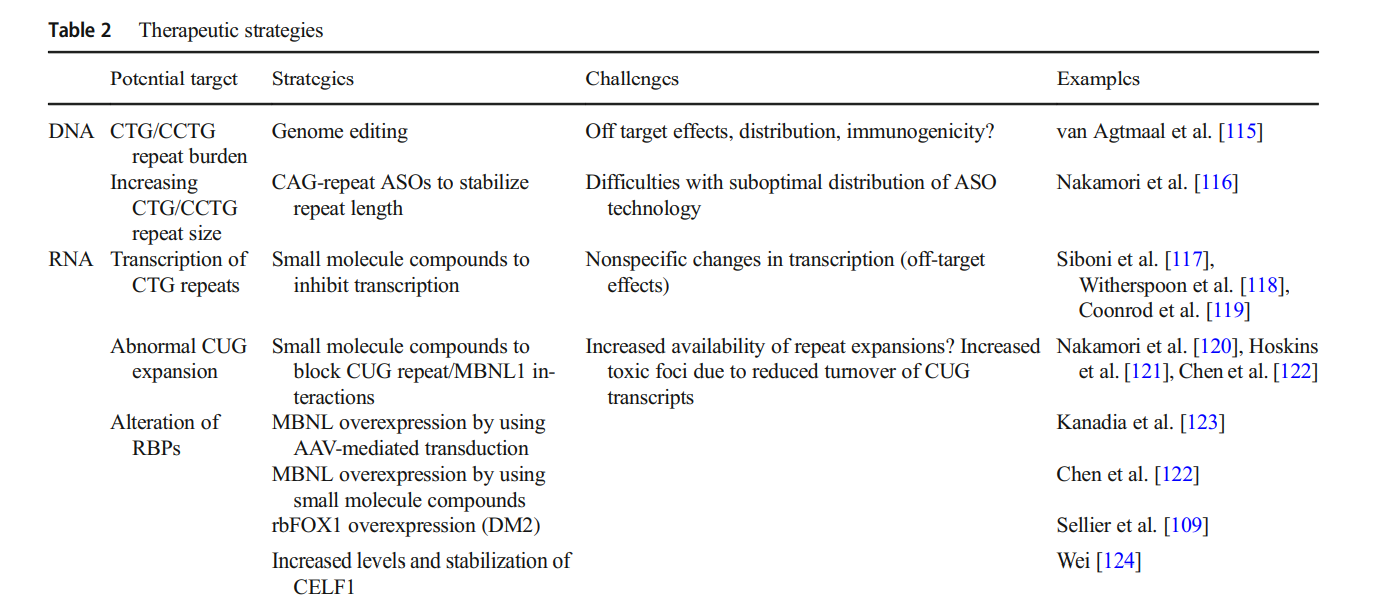
Scientists are exploring gene therapy and molecular interventions to correct genetic mutations or mitigate the effects of toxic RNA. Current areas of research include:
- Genome Editing: Potential to correct repeat expansions.
- Antisense Oligonucleotides (ASO): Designed to target toxic RNA and restore normal RNA splicing.
- Small Molecule Drugs: Targeting RNA toxicity and protein dysfunction.
Challenges and Future Directions
Despite promising advances, several challenges remain, including:
- Effective therapy delivery to muscles and the central nervous system.
- Long-term safety and efficacy of treatments.
- Development of biomarkers for monitoring disease progression and response to therapy.
**Ongoing clinical trials** aim to evaluate these therapies, offering hope for improved treatment outcomes and enhanced quality of life for patients.

| Published | 10/18/2018 |
| Address | https://doi.org/10.1007/s13311-018-00679-z |
| Authors | Samantha LoRusso1 & Benjamin Weiner2 & W. David Arnold1 |











