

اختلال اتوفاژی در دیستروفی عضلانی دوشن (DMD)
دیستروفی عضلانی دوشن (DMD) یک اختلال ژنتیکی است که با ضعف عضلانی پیشرونده به دلیل عدم وجود دیستروفین مشخص میشود. این مطالعه به بررسی اختلال اتوفاژی، یک فرآیند سلولی که پروتئینها و اندامکهای آسیبدیده را تخریب میکند، در عضلات اسکلتی بیماران DMD و مدل موشی mdx میپردازد.
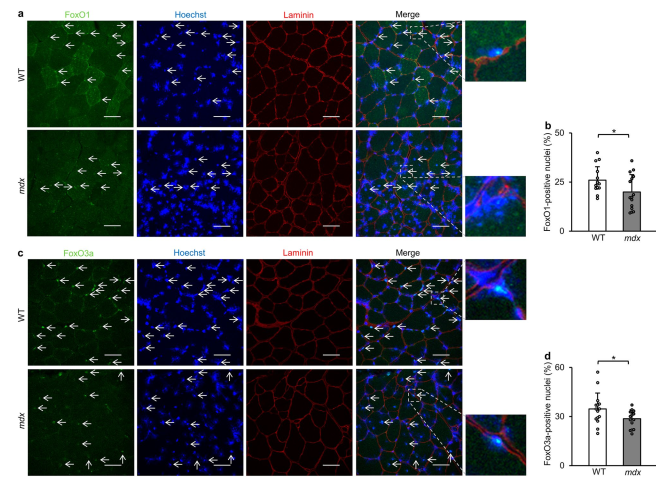
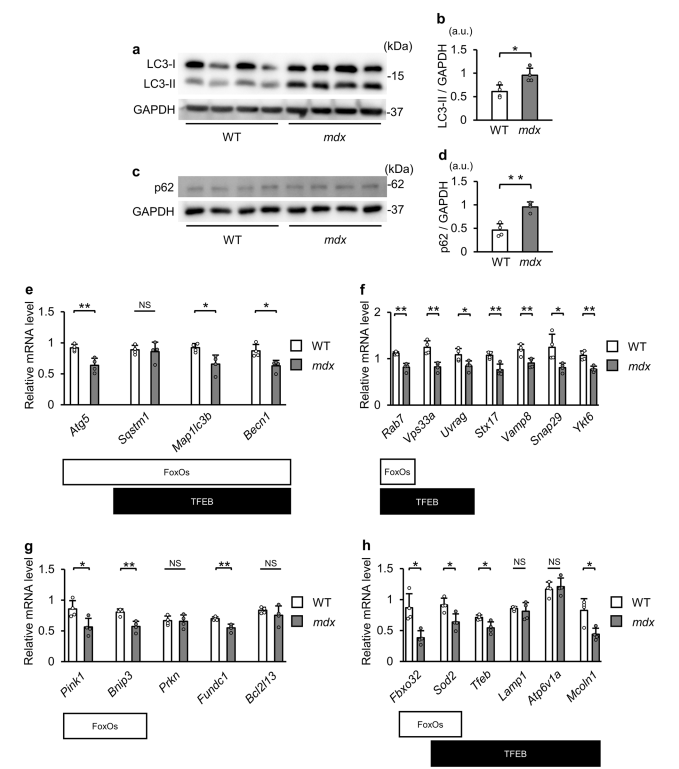
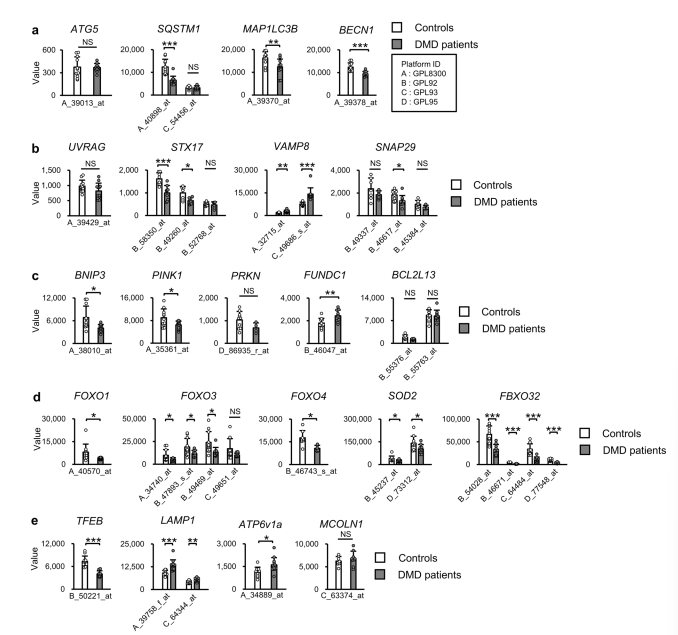
نتایج نشان داد که ژنهای مرتبط با اتوفاژی در ماهیچههای فاقد دیستروفین کاهش مییابند، که عمدتاً به دلیل فسفوریلاسیون و غیرفعال شدن فاکتورهای کلیدی رونویسی مانند FoxO و TFEB است. این فسفوریلاسیون از ورود این عوامل به هسته جلوگیری میکند و توانایی آنها را در ترویج بیان ژنهای مرتبط با اتوفاژی کاهش میدهد.
تجمع اتوفاگوزومها و نقص در اتوفاژی
یافتهها نشان دادند که اختلال در فرآیند اتوفاژی منجر به تجمع اتوفاگوزومها میشود. این نشاندهنده شکست در همجوشی اتوفاگوزوم-لیزوزوم است که یک مرحله مهم در اتوفاژی محسوب میشود.
جالب توجه است که درمانهایی مانند رسوراترول که SIRT1 را فعال میکند و فسفوریلاسیون FoxO و TFEB را کاهش میدهد، میتواند فعالیت اتوفاژیک را بازیابی کرده و عملکرد ماهیچهها را در موشهای mdx بهبود بخشد. این نشان میدهد که هدف قرار دادن اختلال رونویسی اتوفاژی ممکن است یک استراتژی درمانی امیدوارکننده برای کاهش انحطاط عضلانی در DMD باشد.
اهمیت بازیابی عملکرد اتوفاژیک در DMD
این مطالعه بر اهمیت تصحیح سرکوب رونویسی ژنهای مرتبط با اتوفاژی در DMD برای بازگرداندن عملکرد اتوفاژیک مناسب و کاهش آسیب عضلانی تأکید میکند.
تحقیقات آینده باید فاکتورهای رونویسی اضافی و مکانیسمهای اپیژنتیکی مرتبط با این فرآیند را بررسی کنند تا درمانهای مؤثرتری برای DMD توسعه یابند.
| Published | 1/16/2024 |
| Address | https://doi.org/10.1038/s41598-024-51746-9 |
| Authors | Ryuta Nakashima1, Ryusuke Hosoda1, Yuki Tatekoshi1, Naotoshi Iwahara1,2, Yukika Saga1 & Atsushi Kuno |











